Session-4
Aim
- Learn about packages in R
- Use Bioconductor
Packages
Packages are collection of several functions in R.
To use a perticular function from a package, it need to be downloaded and loaded.
Install a package from CRAN
Introduction to Bioconductor
Bioconductor is a collection of more than 1,500 packages for the statistical analysis and comprehension of high-throughput genomic data. Originally developed for microarrays, Bioconductor packages are now used in a wide range of analyses, including bulk and single-cell RNA-seq, ChIP seq, copy number analysis, microarray methylation and classic expression analysis, flow cytometry, and many other domains.
This session introduces the essential of Bioconductor package discovery, installation, and use.
Use Biocondutor
Discovering, installing, and learning how to use Bioconductor packages.
The web site at https://bioconductor.org contains descriptions of all Bioconductor packages, as well as essential reference material for all levels of user.
Application: RNASeq DGE Analysis
RNA sequencing (RNA-seq) has proven as a revolutionary tool since the time it has been introduced. The throughput, accuracy, and resolution of data produced with RNA-seq has been instrumental in the study of Transcriptomics in the last decade (Wang, Gerstein, and Snyder 2009). There are many applications of Transcriptomics data, today we going to discuss Differential Gene Expression (DGE) which is wildly used to study the expression pattern of genes in contrasting samples.
Most of the steps of RNA-seq analysis have become quite mature over the years, especially how to reach a read count table from raw fastq reads obtained from an Illumina sequencing run. We will demonstrate how to process the count table, make a case-control differential expression analysis, and do some downstream functional enrichment analysis.
About the data
For this exercise purpose we will be using the RNA-seq count table from a colorectal cancer study (SRA: SRP029880, collected from recount2 database), only protein coding genes were kept for the faster execution of the analysis.
Get to know the data
Download the data
dir.create("data")
download.file(url = "https://github.com/bionivid-tech/r-workshop-sep-2020/raw/master/data/SRP029880.raw_counts.tsv",
destfile = "data/SRP029880.raw_counts.tsv")
download.file(url = "https://github.com/bionivid-tech/r-workshop-sep-2020/raw/master/data/SRP029880.colData.tsv",
destfile = "data/SRP029880.colData.tsv")Get data into R
Reading tabular in R.
counts <- read.table("data/SRP029880.raw_counts.tsv", sep = "\t", header = T, row.names = 1)
head(counts)## CASE_1 CASE_2 CASE_3 CASE_4 CASE_5 CTRL_1 CTRL_2 CTRL_3 CTRL_4 CTRL_5
## TSPAN6 776426 371725 612244 456147 513335 559544 489653 332084 238516 634115
## TNMD 1483 806 2995 297 1095 4631 1884 4484 1961 3976
## DPM1 364919 274342 248740 371045 325628 211173 123204 113606 67338 198331
## SCYL3 103601 97625 98387 117366 101943 160847 106890 106938 64928 101515
## C1ORF112 90805 59235 61460 108892 44839 52308 34236 38087 18522 58008
## FGR 57081 145623 26800 168519 158882 59723 24483 21608 18654 23682
## width
## TSPAN6 12883
## TNMD 15084
## DPM1 23689
## SCYL3 44637
## C1ORF112 192074
## FGR 23214metadata <- read.table("data/SRP029880.colData.tsv", sep = "\t", header = T, row.names = 1)
metadata## source_name group
## CASE_1 metastasized cancer CASE
## CASE_2 metastasized cancer CASE
## CASE_3 metastasized cancer CASE
## CASE_4 metastasized cancer CASE
## CASE_5 metastasized cancer CASE
## CTRL_1 normal colon CTRL
## CTRL_2 normal colon CTRL
## CTRL_3 normal colon CTRL
## CTRL_4 normal colon CTRL
## CTRL_5 normal colon CTRLNormalization the data
In RNA-Seq, there are different type of normalization methods (such as - CPM, FPKM/RPKM, TPM) and some other method/package specific (Such as in DESeq2 - VST). Today we will calculate CPM and RPKM manually.
CPM
Counts per million (CPM) mapped reads are counts scaled by the number of fragments you sequenced (N) times one million. This unit is related to the FPKM without length normalization and a factor of 10^3 Ref
library(edgeR)
raw_counts <- subset(counts, select = c(-width))
cpm <- cpm.default(raw_counts)
head(cpm)## CASE_1 CASE_2 CASE_3 CASE_4 CASE_5 CTRL_1
## TSPAN6 133.0525426 69.0267115 118.0347011 63.45319990 75.1705899 83.6818109
## TNMD 0.2541349 0.1496685 0.5774069 0.04131475 0.1603471 0.6925826
## DPM1 62.5344859 50.9433750 47.9546579 51.61492360 47.6835767 31.5816791
## SCYL3 17.7536255 18.1282742 18.9680587 16.32642166 14.9280985 24.0552454
## C1ORF112 15.5608340 10.9995218 11.8488915 15.14762970 6.5660321 7.8228489
## FGR 9.7817077 27.0411643 5.1667799 23.44215745 23.2660030 8.9317887
## CTRL_2 CTRL_3 CTRL_4 CTRL_5
## TSPAN6 90.8357832 63.7170750 66.1201154 116.5649088
## TNMD 0.3495018 0.8603467 0.5436178 0.7308802
## DPM1 22.8556382 21.7976235 18.6670761 36.4577954
## SCYL3 19.8292196 20.5182320 17.9989890 18.6607898
## C1ORF112 6.3511382 7.3077662 5.1345687 10.6632034
## FGR 4.5418541 4.1459346 5.1711610 4.3532958RPKM
Reads per kilobase of exon per million reads mapped (RPKM), or the more generic FPKM (substitute reads with fragments) are essentially the same thing. Contrary to some misconceptions, FPKM is not 2 * RPKM if you have paired-end reads. FPKM == RPKM if you have single-end reads, and saying RPKM when you have paired-end reads is just weird, so don’t do it :). Ref
# create a vector of gene lengths
geneLengths <- as.vector(subset(counts, select = c(width)))
# compute rpkm
rpkm <- apply(X = subset(counts, select = c(-width)),
MARGIN = 2,
FUN = function(x) 10^9 * x / geneLengths / sum(as.numeric(x)))
options(scipen=999) # to off scientific notion of numbers
rpkm <- do.call(cbind, rpkm)
options(scipen=0, digits=7) # on again back
head(rpkm)## width width width width width width
## TSPAN6 10.32776081 5.357968757 9.16205085 4.925343468 5.83486687 6.49552207
## TNMD 0.01684798 0.009922336 0.03827943 0.002738979 0.01063028 0.04591505
## DPM1 2.63981113 2.150507621 2.02434285 2.178856161 2.01289952 1.33317907
## SCYL3 0.39773339 0.406126625 0.42494027 0.365759833 0.33443328 0.53890820
## C1ORF112 0.08101479 0.057267105 0.06168920 0.078863509 0.03418491 0.04072831
## FGR 0.42137106 1.164864492 0.22257172 1.009828442 1.00224016 0.38475871
## width width width width
## TSPAN6 7.05082537 4.94582589 5.13235390 9.04796312
## TNMD 0.02317037 0.05703704 0.03603937 0.04845400
## DPM1 0.96482073 0.92015803 0.78800608 1.53901792
## SCYL3 0.44423280 0.45966871 0.40323026 0.41805654
## C1ORF112 0.03306610 0.03804662 0.02673224 0.05551612
## FGR 0.19565151 0.17859631 0.22276045 0.18752890Quality check of samples
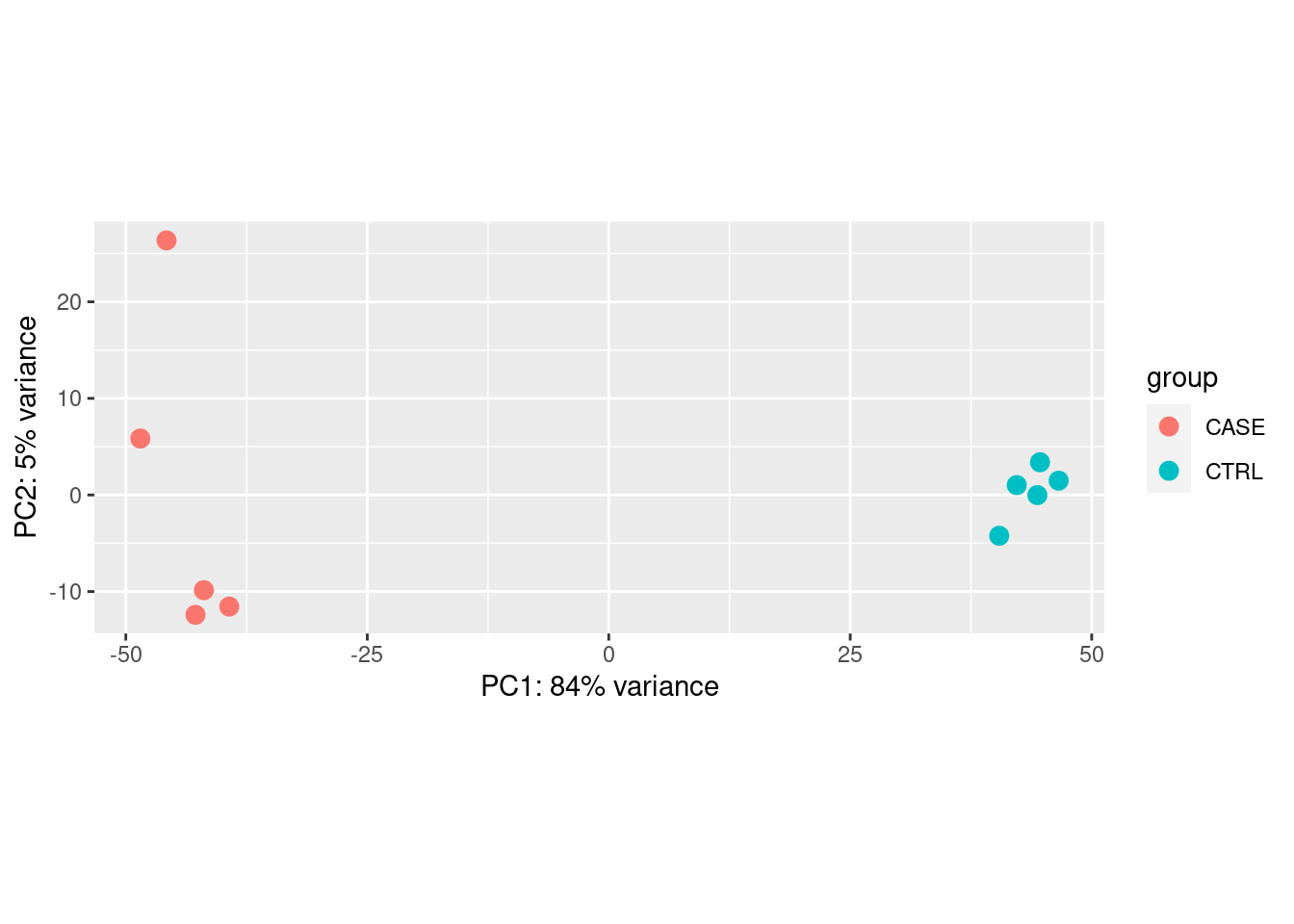
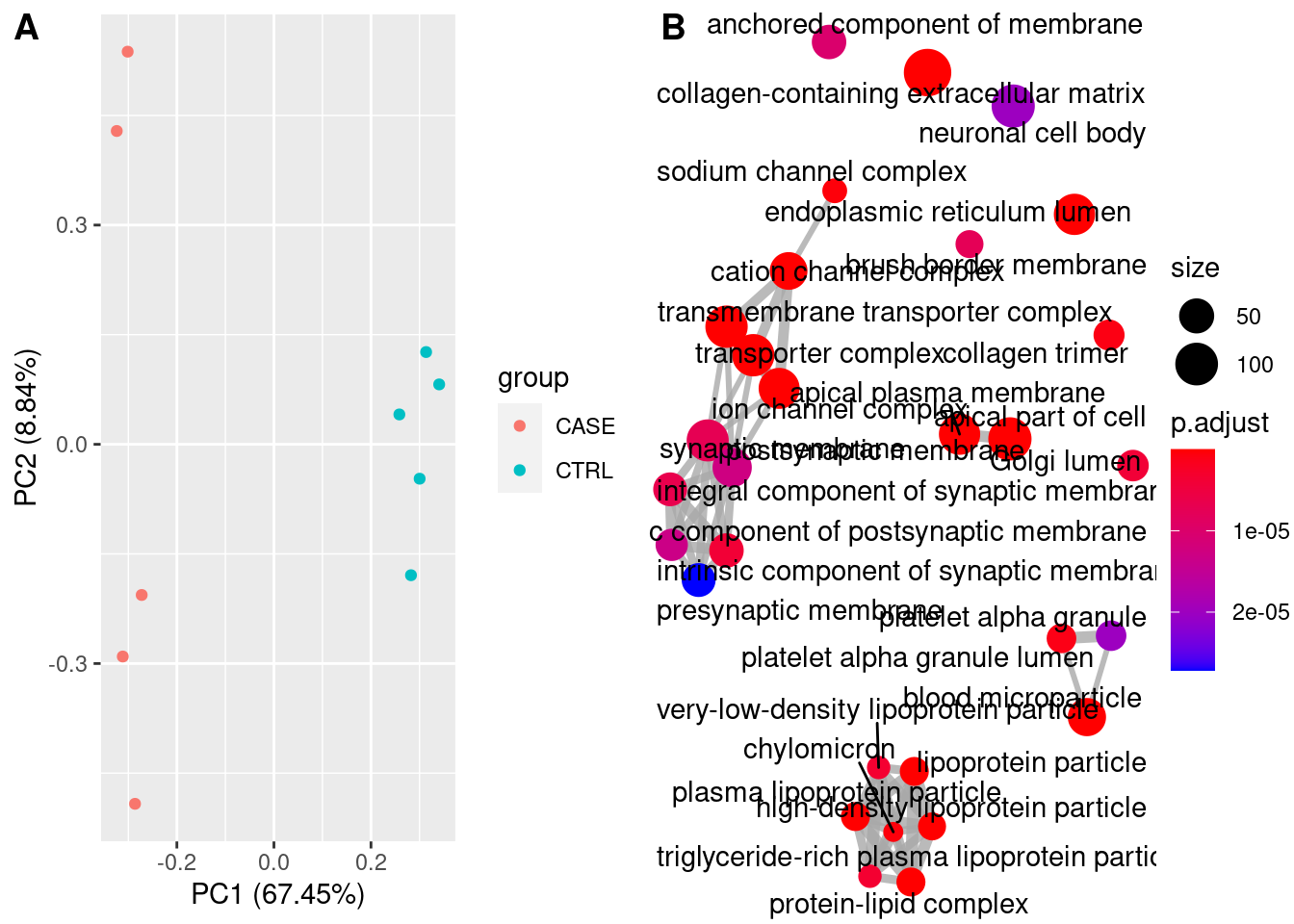
For quality check of samples we can do several studies such as PCA and Correlation.
PCA
Principal Compound analysis on samples.
#transpose the matrix
cpm_t <- t(cpm)
# transform the counts to log2 scale
cpm_log <- log2(cpm_t + 1)
# calculate pca
pca <- prcomp(cpm_log)
summary(pca)## Importance of components:
## PC1 PC2 PC3 PC4 PC5 PC6
## Standard deviation 80.0323 28.96809 23.83710 22.75690 18.86937 16.30932
## Proportion of Variance 0.6745 0.08837 0.05984 0.05453 0.03749 0.02801
## Cumulative Proportion 0.6745 0.76286 0.82270 0.87723 0.91473 0.94274
## PC7 PC8 PC9 PC10
## Standard deviation 15.39922 14.21772 10.223 1.039e-13
## Proportion of Variance 0.02497 0.02129 0.011 0.000e+00
## Cumulative Proportion 0.96771 0.98900 1.000 1.000e+00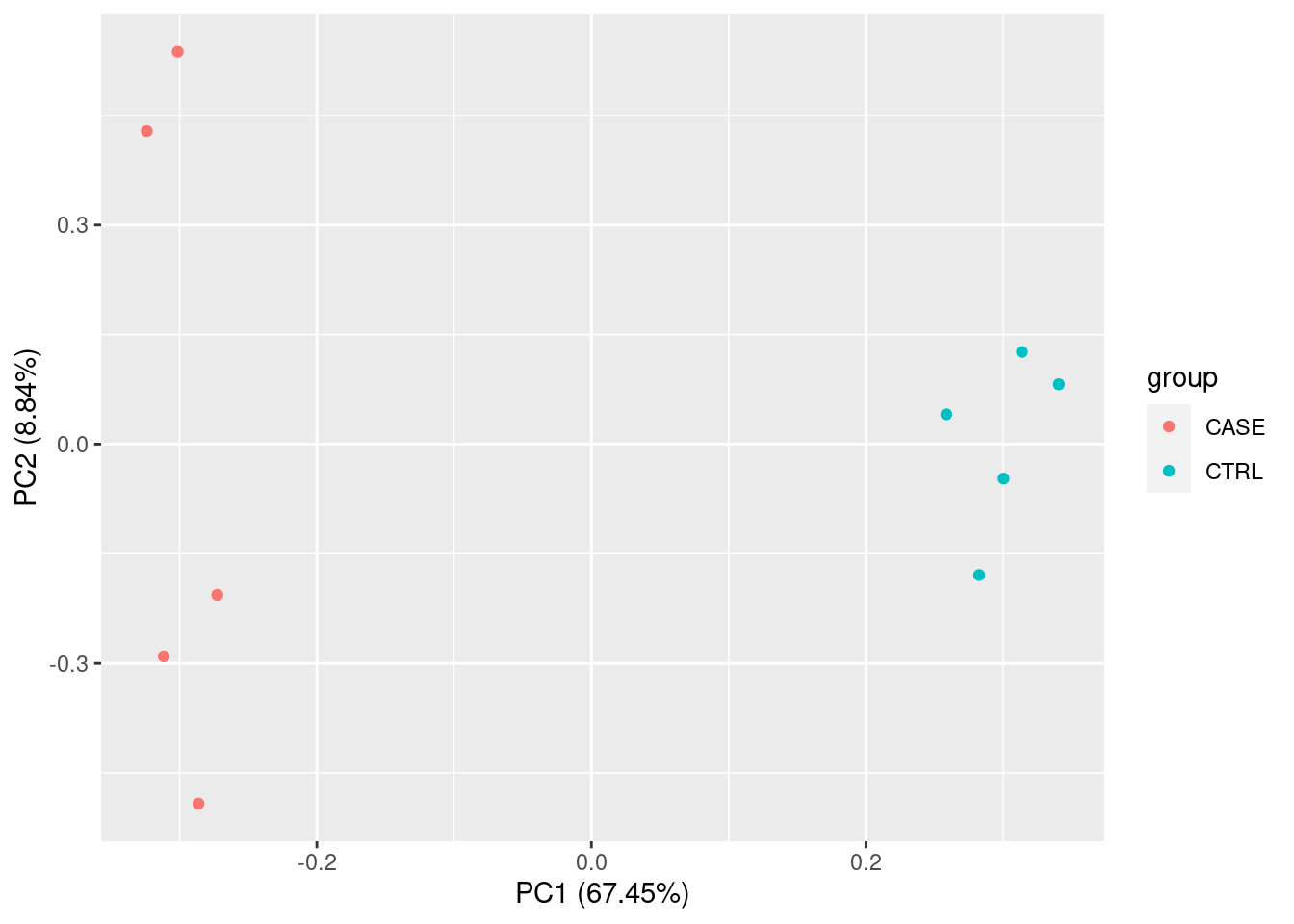
Correlation
Sample to sample relation.
| CASE_1 | CASE_2 | CASE_3 | CASE_4 | CASE_5 | CTRL_1 | CTRL_2 | CTRL_3 | CTRL_4 | CTRL_5 | |
|---|---|---|---|---|---|---|---|---|---|---|
| CASE_1 | 1.0000000 | 0.8932785 | 0.9848753 | 0.9439505 | 0.8750504 | 0.5283715 | 0.5171387 | 0.5191767 | 0.5132126 | 0.5256384 |
| CASE_2 | 0.8932785 | 1.0000000 | 0.8586247 | 0.9635875 | 0.9324452 | 0.7517934 | 0.7263228 | 0.7372209 | 0.7267303 | 0.7427978 |
| CASE_3 | 0.9848753 | 0.8586247 | 1.0000000 | 0.9122808 | 0.8361842 | 0.4780616 | 0.4652480 | 0.4683691 | 0.4628397 | 0.4757110 |
| CASE_4 | 0.9439505 | 0.9635875 | 0.9122808 | 1.0000000 | 0.9461943 | 0.6953668 | 0.6812613 | 0.6831496 | 0.6799802 | 0.6980752 |
| CASE_5 | 0.8750504 | 0.9324452 | 0.8361842 | 0.9461943 | 1.0000000 | 0.7018531 | 0.6789861 | 0.6834173 | 0.6756385 | 0.6956374 |
| CTRL_1 | 0.5283715 | 0.7517934 | 0.4780616 | 0.6953668 | 0.7018531 | 1.0000000 | 0.9712296 | 0.9886670 | 0.9784986 | 0.9803462 |
| CTRL_2 | 0.5171387 | 0.7263228 | 0.4652480 | 0.6812613 | 0.6789861 | 0.9712296 | 1.0000000 | 0.9817525 | 0.9951188 | 0.9782734 |
| CTRL_3 | 0.5191767 | 0.7372209 | 0.4683691 | 0.6831496 | 0.6834173 | 0.9886670 | 0.9817525 | 1.0000000 | 0.9871216 | 0.9849108 |
| CTRL_4 | 0.5132126 | 0.7267303 | 0.4628397 | 0.6799802 | 0.6756385 | 0.9784986 | 0.9951188 | 0.9871216 | 1.0000000 | 0.9837302 |
| CTRL_5 | 0.5256384 | 0.7427978 | 0.4757110 | 0.6980752 | 0.6956374 | 0.9803462 | 0.9782734 | 0.9849108 | 0.9837302 | 1.0000000 |
Correlation Plot
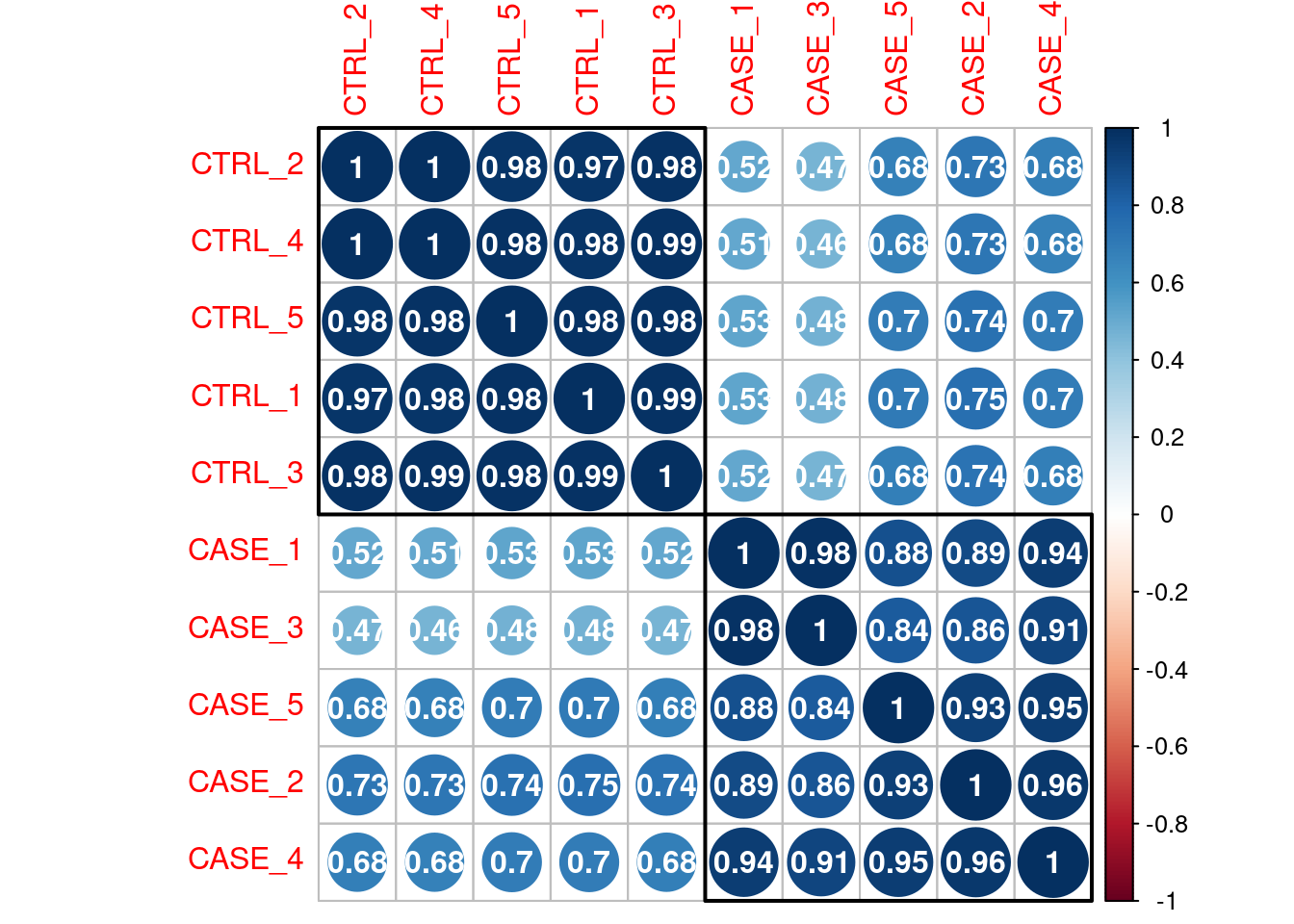
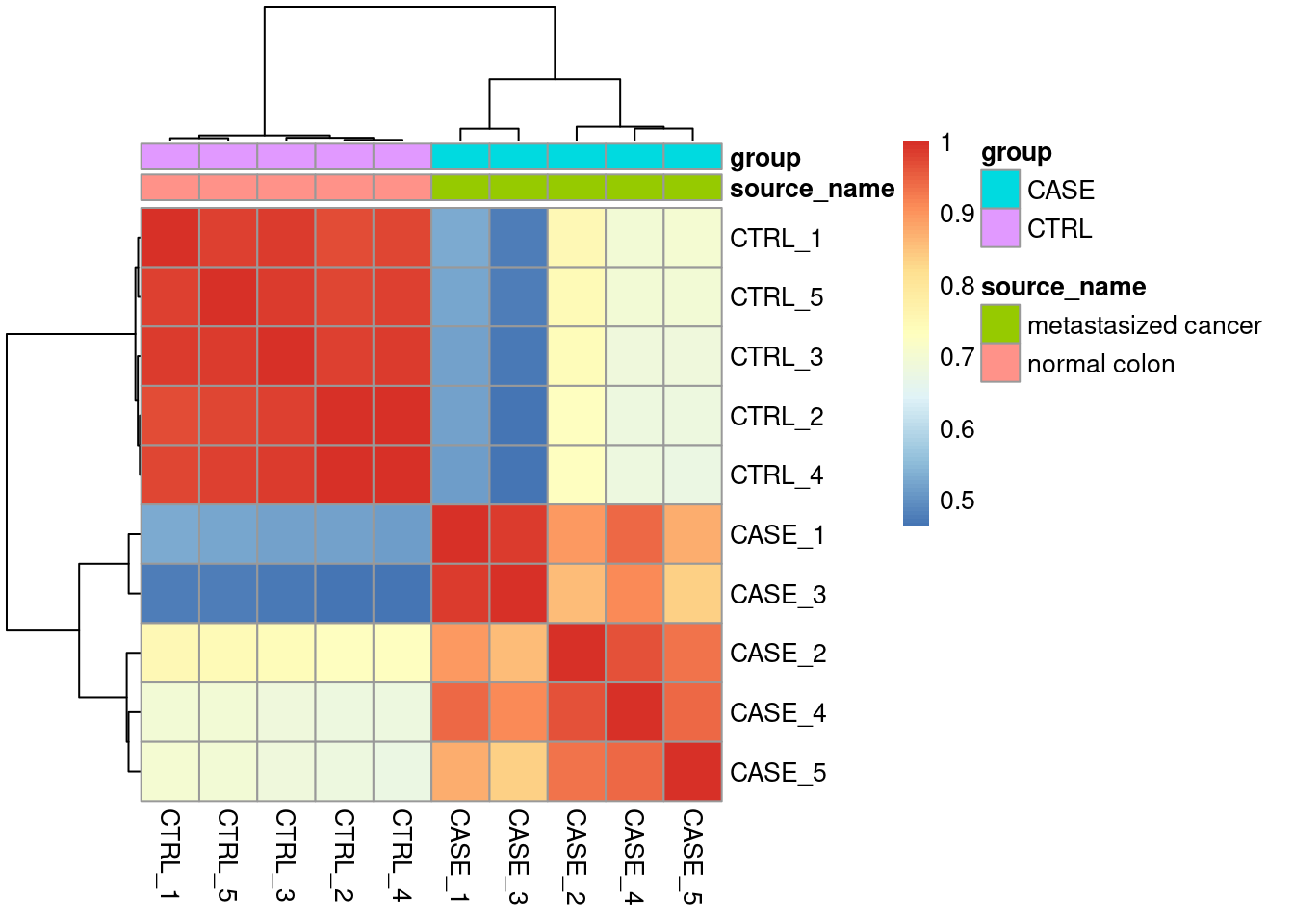
DEG Analsysis
Make the data ready.
#remove the 'width' column
countdata <- as.matrix(subset(counts, select = c(-width)))
head(countdata)## CASE_1 CASE_2 CASE_3 CASE_4 CASE_5 CTRL_1 CTRL_2 CTRL_3 CTRL_4 CTRL_5
## TSPAN6 776426 371725 612244 456147 513335 559544 489653 332084 238516 634115
## TNMD 1483 806 2995 297 1095 4631 1884 4484 1961 3976
## DPM1 364919 274342 248740 371045 325628 211173 123204 113606 67338 198331
## SCYL3 103601 97625 98387 117366 101943 160847 106890 106938 64928 101515
## C1ORF112 90805 59235 61460 108892 44839 52308 34236 38087 18522 58008
## FGR 57081 145623 26800 168519 158882 59723 24483 21608 18654 23682## source_name group
## CASE_1 metastasized cancer CASE
## CASE_2 metastasized cancer CASE
## CASE_3 metastasized cancer CASE
## CASE_4 metastasized cancer CASE
## CASE_5 metastasized cancer CASE
## CTRL_1 normal colon CTRL
## CTRL_2 normal colon CTRL
## CTRL_3 normal colon CTRL
## CTRL_4 normal colon CTRL
## CTRL_5 normal colon CTRLLoad the data into DESeq2 Object and design the experiment
library(DESeq2)
dds <- DESeqDataSetFromMatrix(countData = countdata,
colData = metadata,
design = ~ group) # experiment design
dds## class: DESeqDataSet
## dim: 19719 10
## metadata(1): version
## assays(1): counts
## rownames(19719): TSPAN6 TNMD ... MYOCOS HSFX3
## rowData names(0):
## colnames(10): CASE_1 CASE_2 ... CTRL_4 CTRL_5
## colData names(2): source_name groupRemove genes with almost no information in any of the given samples.
#For each gene, we count the total number of reads for that gene in all samples
#and remove those that don't have at least 1 read.
dds <- dds[ rowSums(counts(dds)) > 1, ]Run the analysis
## estimating size factors## estimating dispersions## gene-wise dispersion estimates## mean-dispersion relationship## final dispersion estimates## fitting model and testing## class: DESeqDataSet
## dim: 19097 10
## metadata(1): version
## assays(4): counts mu H cooks
## rownames(19097): TSPAN6 TNMD ... MYOCOS HSFX3
## rowData names(22): baseMean baseVar ... deviance maxCooks
## colnames(10): CASE_1 CASE_2 ... CTRL_4 CTRL_5
## colData names(3): source_name group sizeFactorExplore results
#compute the contrast for the 'group' variable where 'CTRL' samples are used as the control group.
res = results(dds, contrast = c("group", 'CASE', 'CTRL'))
head(res)## log2 fold change (MLE): group CASE vs CTRL
## Wald test p-value: group CASE vs CTRL
## DataFrame with 6 rows and 6 columns
## baseMean log2FoldChange lfcSE stat pvalue padj
## <numeric> <numeric> <numeric> <numeric> <numeric> <numeric>
## TSPAN6 491987.63 0.0772416 0.283184 0.272761 7.85037e-01 8.68555e-01
## TNMD 2466.25 -1.4474155 0.631826 -2.290846 2.19723e-02 5.65051e-02
## DPM1 216809.25 0.9388739 0.200290 4.687581 2.76453e-06 2.28302e-05
## SCYL3 104400.07 -0.2843211 0.113732 -2.499919 1.24222e-02 3.54271e-02
## C1ORF112 53848.01 0.6299092 0.271324 2.321609 2.02540e-02 5.28672e-02
## FGR 63201.42 1.6468890 0.410016 4.016646 5.90324e-05 3.56989e-04## log2 fold change (MLE): group CASE vs CTRL
## Wald test p-value: group CASE vs CTRL
## DataFrame with 19097 rows and 6 columns
## baseMean log2FoldChange lfcSE stat pvalue
## <numeric> <numeric> <numeric> <numeric> <numeric>
## CYP2E1 4829889 9.36024 0.215223 43.4909 0.00000e+00
## FCGBP 10349993 -7.57579 0.186433 -40.6355 0.00000e+00
## ASGR2 426422 8.01830 0.216207 37.0863 4.67898e-301
## GCKR 100183 7.82841 0.233376 33.5442 1.09479e-246
## APOA5 438054 10.20248 0.312503 32.6477 8.64906e-234
## ... ... ... ... ... ...
## CCDC195 20.4981 -0.215607 2.89255 -0.0745386 NA
## SPEM3 23.6370 -22.154765 3.02785 -7.3170030 NA
## AC022167.5 21.8451 -2.056240 2.89545 -0.7101618 NA
## BX276092.9 29.9636 0.407326 2.89048 0.1409199 NA
## ETDC 22.5675 -1.795274 2.89421 -0.6202983 NA
## padj
## <numeric>
## CYP2E1 0.00000e+00
## FCGBP 0.00000e+00
## ASGR2 2.87741e-297
## GCKR 5.04945e-243
## APOA5 3.19133e-230
## ... ...
## CCDC195 NA
## SPEM3 NA
## AC022167.5 NA
## BX276092.9 NA
## ETDC NA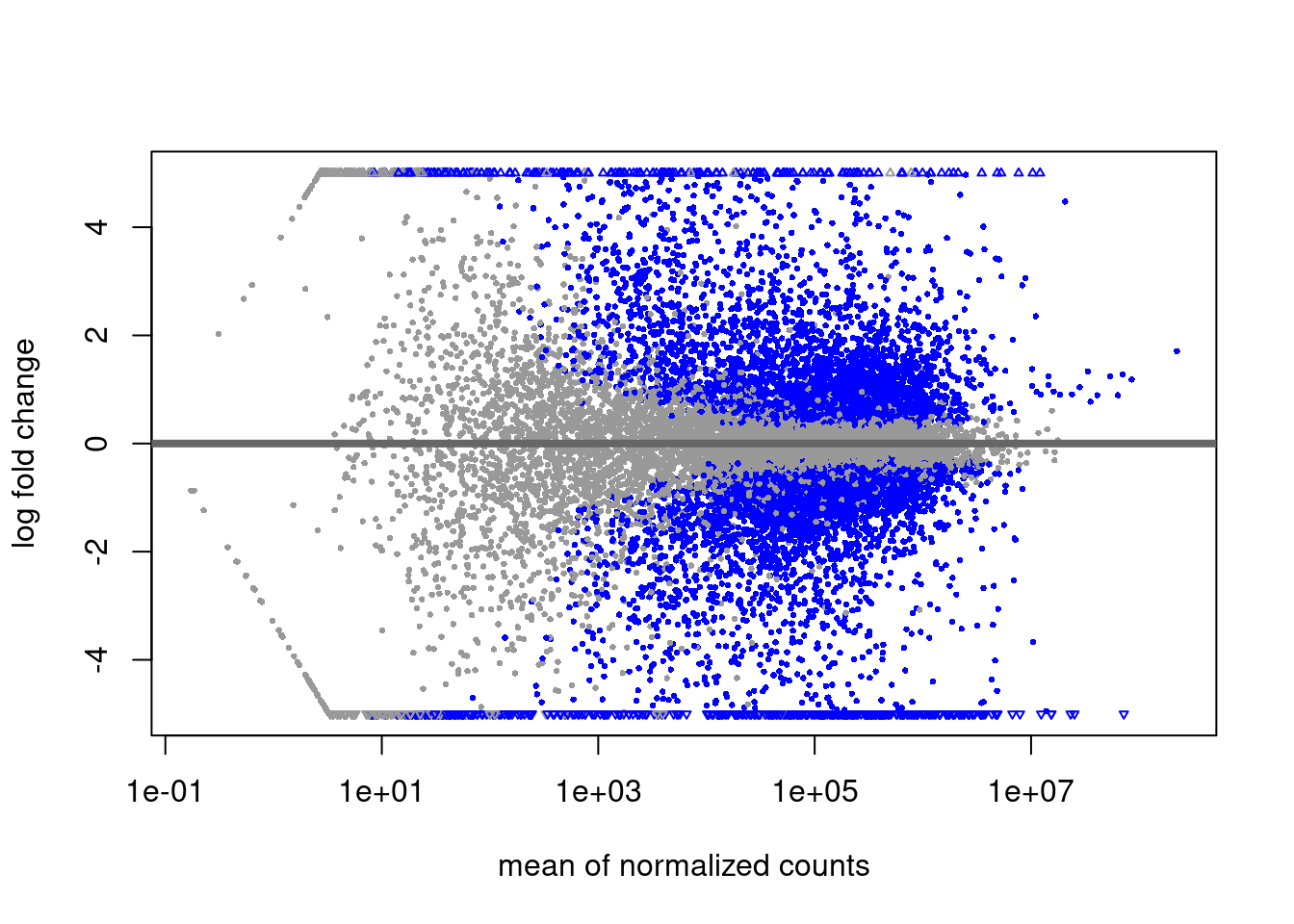
P-value distribution
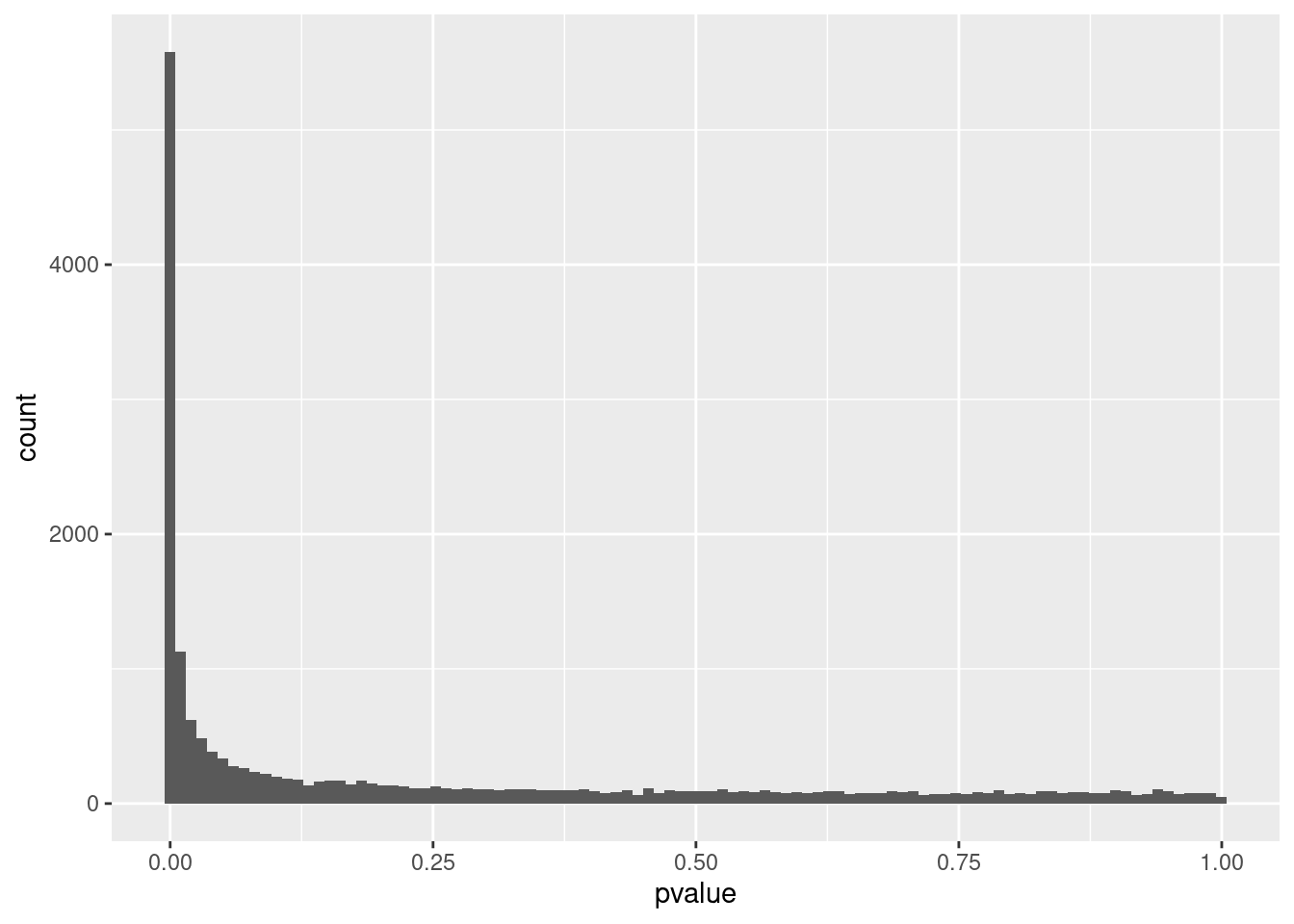
Box plot
For deseq normalized
par(mfrow = c(1, 2))
boxplot(log(countdata),
col = as.numeric(as.factor(metadata$group)),
main = 'Raw Counts')
boxplot(log(counts(dds, normalized = TRUE)),
col = as.numeric(as.factor(metadata$group)),
main = 'Normalized Counts')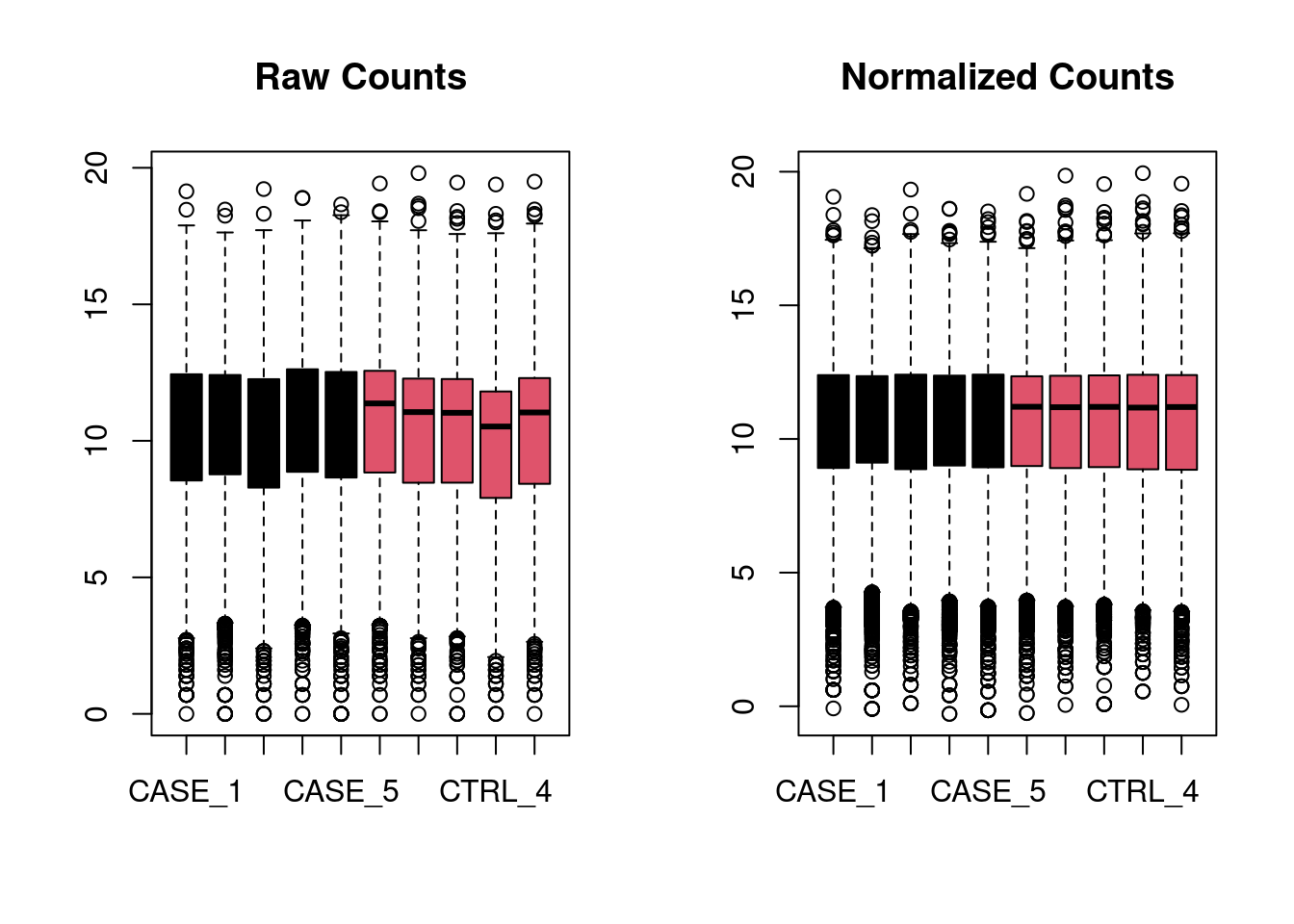
DGE Volcano plot
library(EnhancedVolcano)
EnhancedVolcano(res,
lab = rownames(res),
x = 'log2FoldChange',
y = 'pvalue',
title = "CASE vs CTRL",
pCutoff = 0.05,
FCcutoff = 2,
pointSize = 3.0,
labSize = 3.0)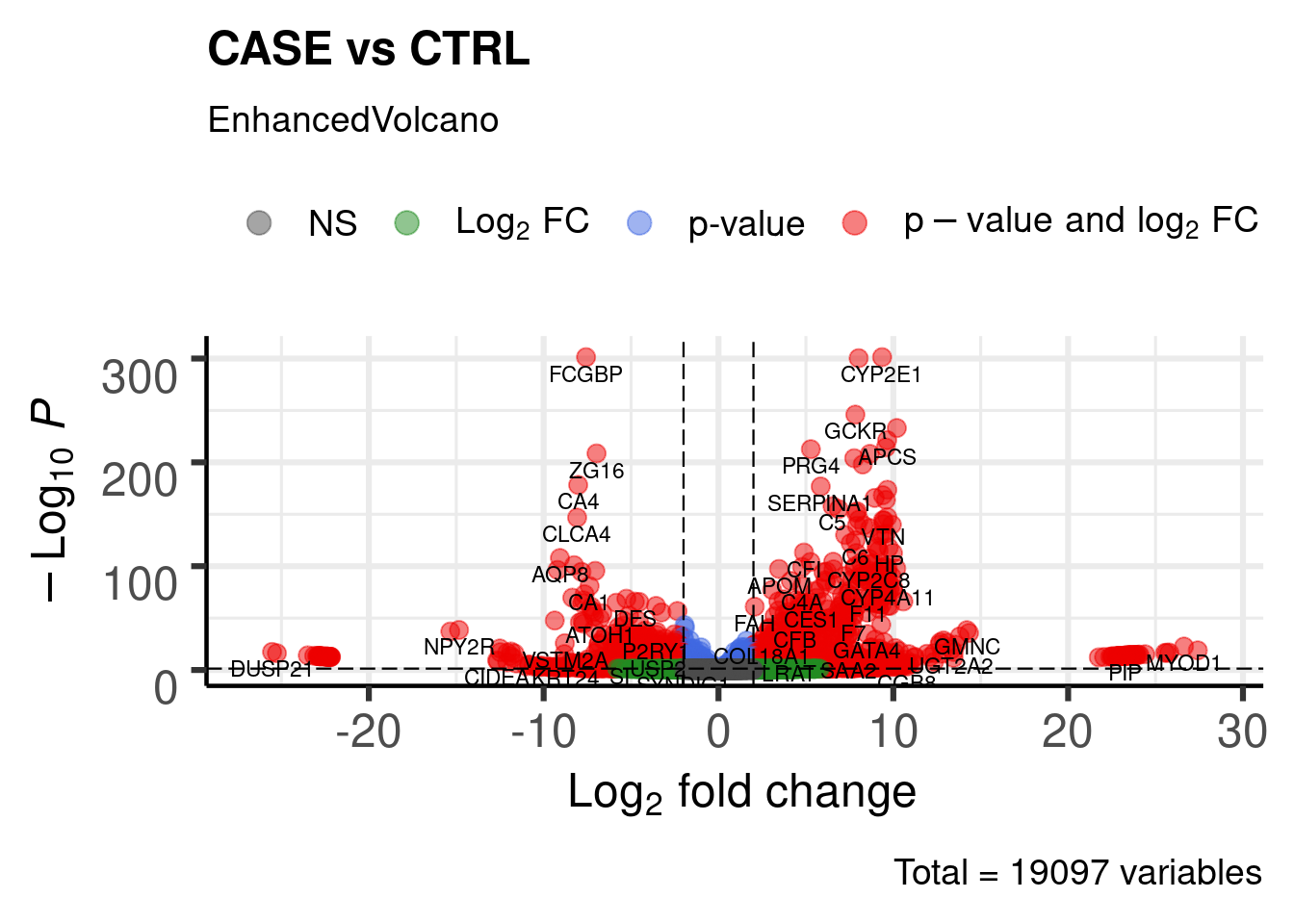
Extract results
Extract the DGE to make heatmap
library(dplyr)
library(tibble)
library(magrittr)
res_up <- res %>% as.data.frame() %>%
rownames_to_column('gene') %>%
filter(log2FoldChange >= 2) %>%
mutate(reg_type = "Up regulation") %>%
column_to_rownames('gene')
head(res_up)## baseMean log2FoldChange lfcSE stat pvalue padj
## CYP2E1 4829888.9 9.360244 0.2152232 43.49088 0.000000e+00 0.000000e+00
## ASGR2 426421.7 8.018301 0.2162068 37.08626 4.678977e-301 2.877415e-297
## GCKR 100182.7 7.828413 0.2333762 33.54417 1.094790e-246 5.049446e-243
## APOA5 438054.0 10.202485 0.3125028 32.64766 8.649058e-234 3.191329e-230
## APCS 899875.4 9.649894 0.3033213 31.81410 4.131410e-222 1.270340e-218
## APOC4 152415.6 9.569025 0.3057277 31.29917 4.788978e-215 1.262169e-211
## reg_type
## CYP2E1 Up regulation
## ASGR2 Up regulation
## GCKR Up regulation
## APOA5 Up regulation
## APCS Up regulation
## APOC4 Up regulationres_down <- res %>% as.data.frame() %>%
rownames_to_column('gene') %>%
filter(log2FoldChange <= -2) %>%
mutate(reg_type = "Down regulation") %>%
column_to_rownames('gene')
head(res_down)## baseMean log2FoldChange lfcSE stat pvalue
## FCGBP 10349993.2 -7.575787 0.1864329 -40.63545 0.000000e+00
## ZG16 2122231.5 -6.974906 0.2258976 -30.87641 2.477314e-209
## CA4 842822.2 -8.025380 0.2813713 -28.52238 6.184050e-179
## CLCA4 1918679.3 -8.085007 0.3125788 -25.86550 1.628578e-147
## AQP8 1741004.4 -9.070041 0.4097381 -22.13619 1.417134e-108
## TMIGD1 325850.3 -8.257870 0.3857252 -21.40869 1.108936e-101
## padj reg_type
## FCGBP 0.000000e+00 Down regulation
## ZG16 5.078219e-206 Down regulation
## CA4 8.776118e-176 Down regulation
## CLCA4 1.251901e-144 Down regulation
## AQP8 5.683630e-106 Down regulation
## TMIGD1 3.860143e-99 Down regulationres_filt <- rbind(res_up, res_down)
# get up and down genes
res_filt_genes <- rownames(res_filt)
res_filt_genes[1:10]## [1] "CYP2E1" "ASGR2" "GCKR" "APOA5" "APCS" "APOC4" "PRG4" "ITIH3"
## [9] "A1BG" "AADAC"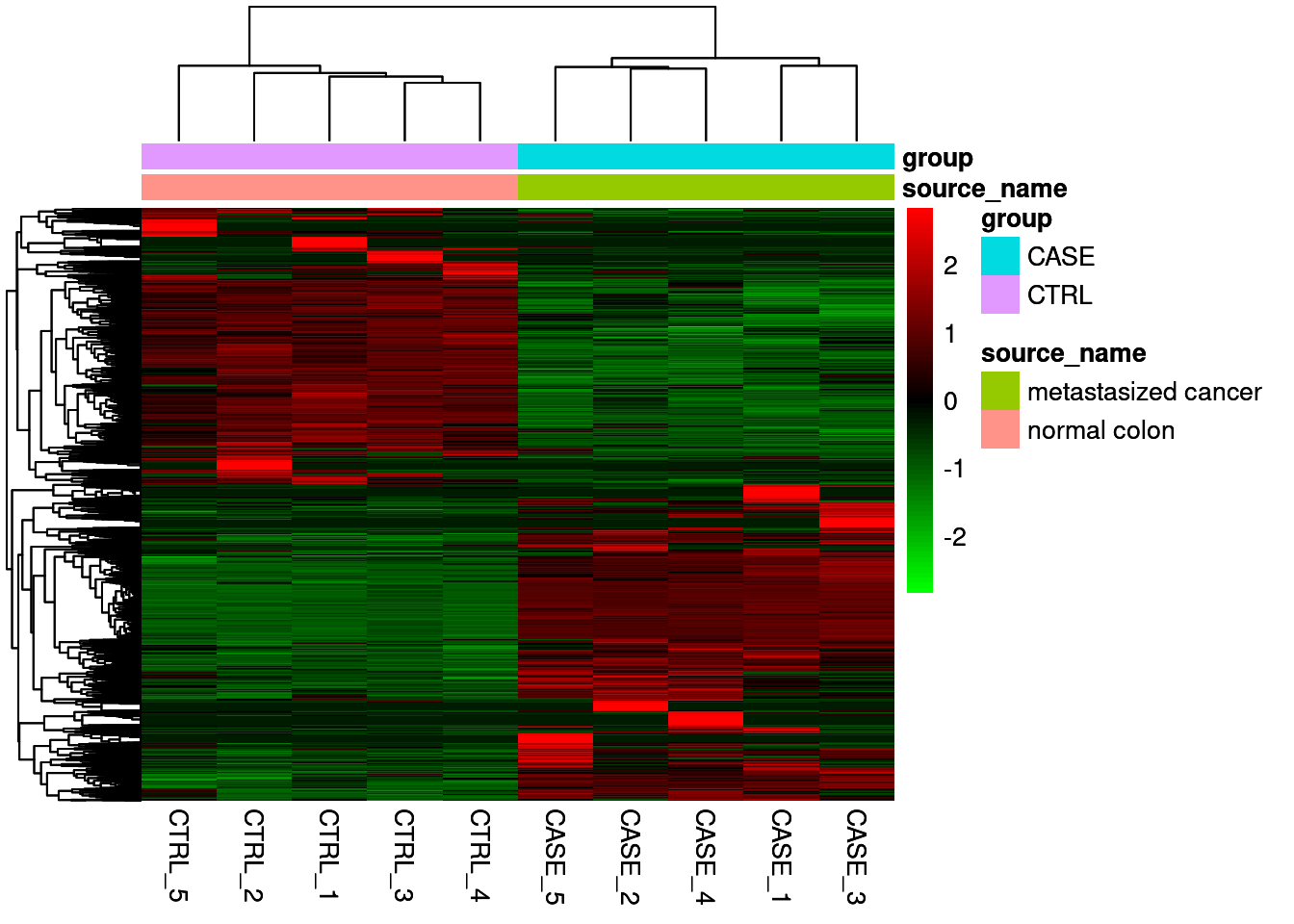
Downstream analaysis
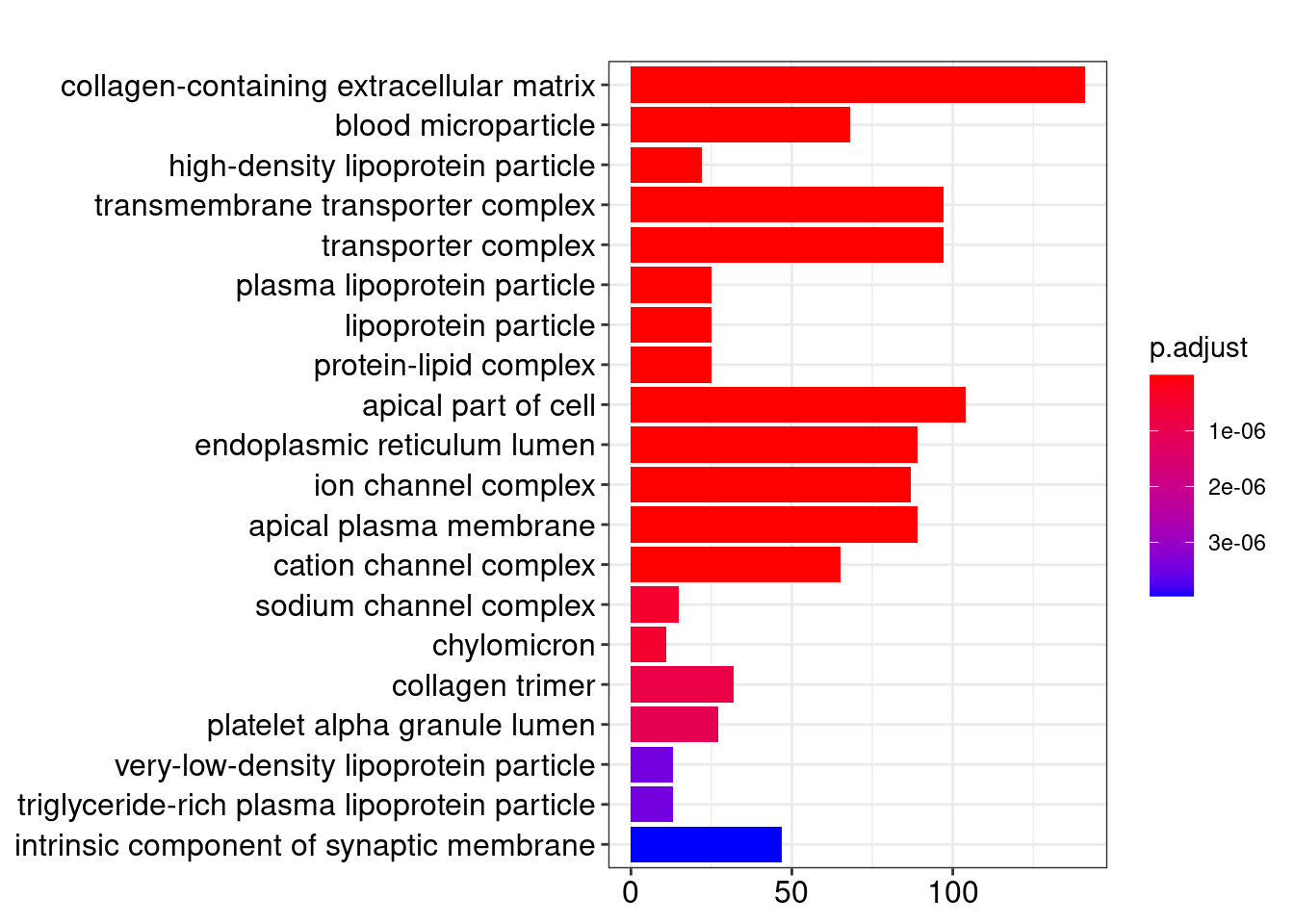
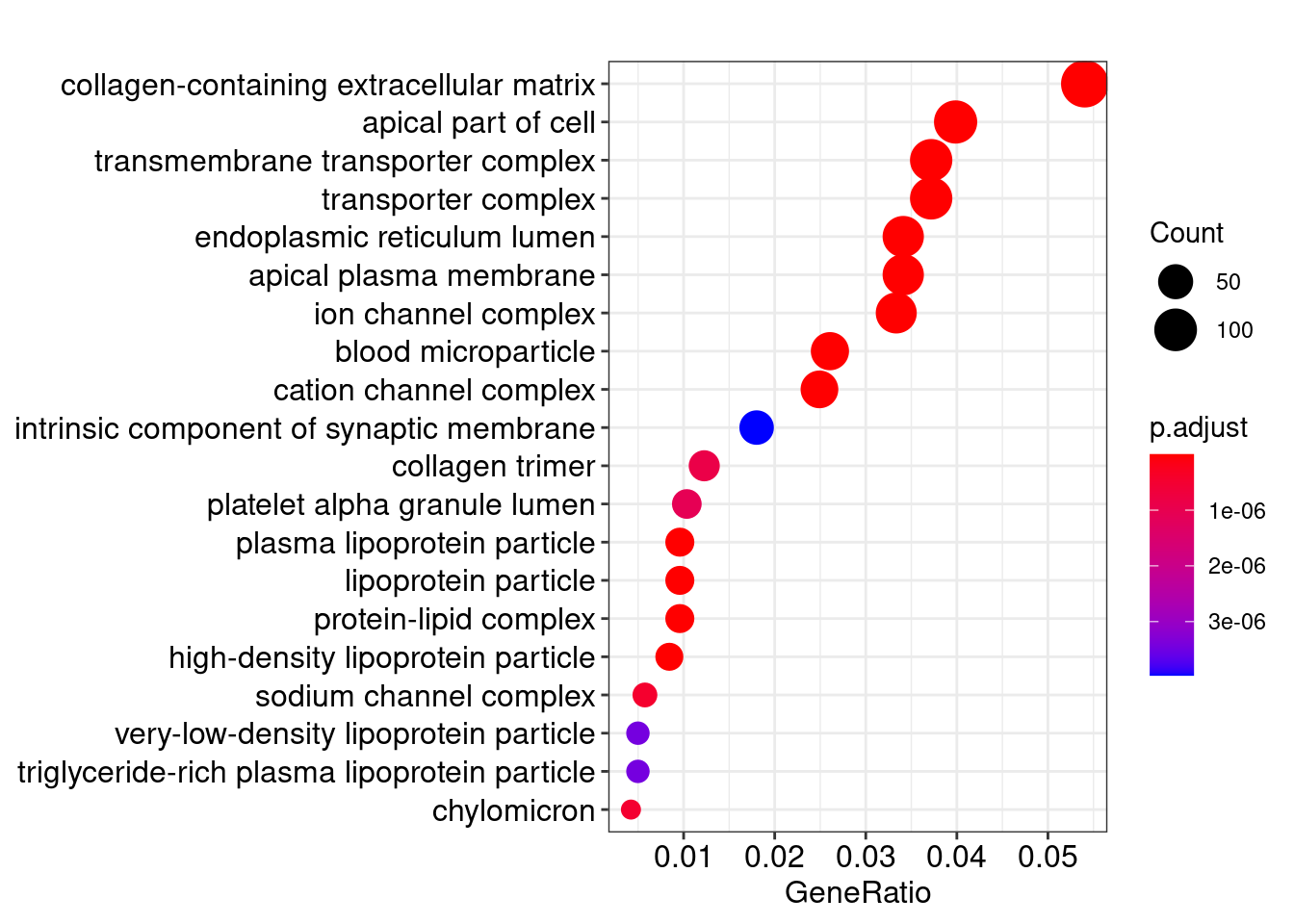
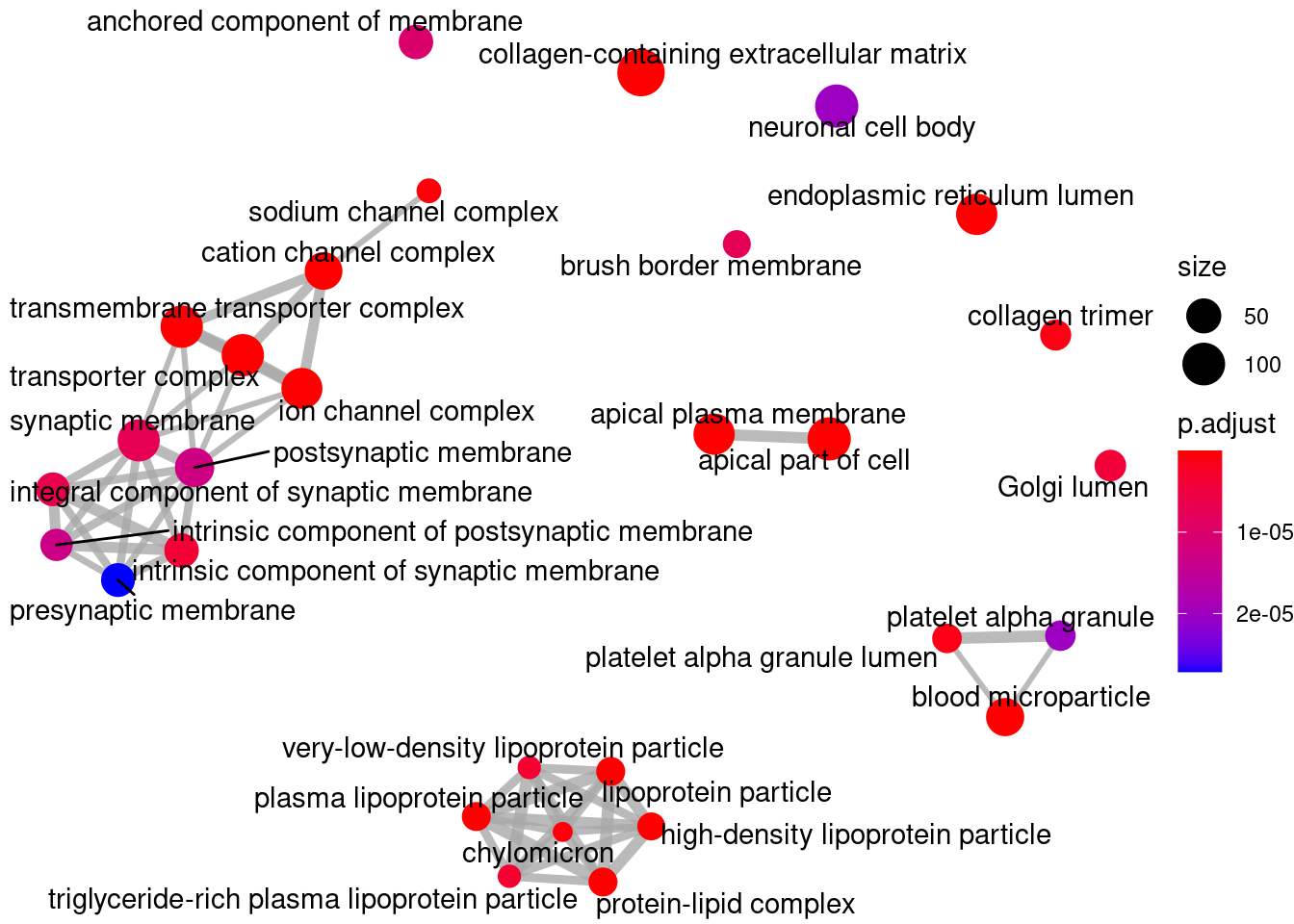
Gene Ontology
library(clusterProfiler)
library(org.Hs.eg.db)
ego <- enrichGO(gene = res_filt_genes,
OrgDb = org.Hs.eg.db,
keyType = "SYMBOL",
ont = "CC",
pvalueCutoff = 0.01,
qvalueCutoff = 0.05)
knitr::kable(head(ego@result))| ID | Description | GeneRatio | BgRatio | pvalue | p.adjust | qvalue | geneID | Count | |
|---|---|---|---|---|---|---|---|---|---|
| GO:0062023 | GO:0062023 | collagen-containing extracellular matrix | 141/2609 | 406/19717 | 0 | 0 | 0 | APCS/PRG4/A1BG/SERPINA1/HPX/SERPINA5/ITIH4/VTN/APOC3/APOA1/ORM2/APOH/PLG/ITIH2/F2/SERPINA3/SERPINF2/ITIH1/F9/AMBP/FGA/ORM1/AHSG/FGG/SERPINC1/FGB/SERPING1/CDH2/F7/F12/FN1/AGT/MST1/INHBE/COL18A1/CLU/HRG/KNG1/ANG/PZP/ADAMTS2/THBS2/APOE/SERPINE1/MBL2/COL10A1/BGN/COL6A6/TGM2/SPP2/SMOC1/COL4A1/AZGP1/ASPN/CTHRC1/CFP/SDC2/USH2A/TIMP1/ANXA8/ADAMTS4/TGFB3/COL4A2/COL1A1/COL26A1/TGFBI/COMP/SULF1/APOA4/COL11A1/SEMA7A/COL25A1/MATN3/MMP9/VWA2/MMP8/DEFA1/BMP7/CBLN4/WNT2/PRTN3/LAMB4/SOST/WNT8A/S100A7/PRG3/ADAMTS20/COL11A2/PRSS1/DEFA1B/ZG16/ADAMDEC1/COL4A6/FGF9/LGALS4/CMA1/MMP28/MFAP5/LAMA1/WNT2B/RBP3/EFNA5/FGF10/LTBP4/EDIL3/OGN/HAPLN1/HMCN2/CTSG/FBLN1/PTPRZ1/TPSAB1/CLEC3B/SFRP1/PTN/SPARCL1/MYOC/FBN2/MATN2/COL9A2/VIT/TPSB2/DPT/APLP1/ADIPOQ/COL19A1/TINAG/SFRP2/CPA3/NPPA/COL28A1/THBS4/SEMA3B/COL9A1/KRT1/VWC2/ZP1/RTBDN/TGM4/MUC17/COL6A5 | 141 |
| GO:0072562 | GO:0072562 | blood microparticle | 68/2609 | 147/19717 | 0 | 0 | 0 | APCS/A1BG/HPX/ITIH4/VTN/APOA1/C9/HP/HPR/ORM2/APOA2/C4B/CPN2/PLG/C4BPA/C8A/ITIH2/F2/SERPINA3/SERPINF2/GC/ITIH1/AMBP/C4A/TF/CP/FCN2/FGA/ORM1/AHSG/FGG/SERPINC1/FGB/CFHR1/SERPING1/C3/PON1/ALB/CD5L/CFHR3/AFM/FN1/CFB/AGT/C8G/C1R/CLU/HRG/KNG1/PZP/PROS1/C1S/CFH/APOE/BCHE/ANGPTL4/FCN3/HSPA6/APOA4/HBE1/PRSS1/JCHAIN/ACTG2/GSN/HBA2/HBA1/HBB/KRT1 | 68 |
| GO:0034364 | GO:0034364 | high-density lipoprotein particle | 22/2609 | 26/19717 | 0 | 0 | 0 | APOA5/APOC4/APOC3/APOC2/APOA1/APOB/HPR/APOC1/APOA2/APOH/APOM/APOF/PON1/LIPC/SAA4/CLU/LCAT/APOE/SAA1/SAA2/CETP/APOA4 | 22 |
| GO:1902495 | GO:1902495 | transmembrane transporter complex | 97/2609 | 324/19717 | 0 | 0 | 0 | KCNJ8/ABCG5/CHRNA4/TRPC5/ABCC9/OLFM2/ABCG8/CACNG1/KCNH4/FXYD2/CACNG8/CACNG4/GABRP/KCNE1B/KCNG4/CHRNA6/GLRA1/GABRR3/ATP1B4/KCNA10/CHRNB3/CHRND/ATP12A/GABRA6/BEST2/BEST4/SCNN1B/CNGA3/LRRC26/ATP1A2/GRIK3/KCNIP4/CHRNA3/TRPM4/HTR3B/OLFM3/CACNA2D2/SLC9A1/KCNH1/GRIK5/CNGB1/CLIC5/SCN3A/KCNK2/GLRA4/CASQ2/PLN/CACNA1A/GRIN1/KCNS3/SCN2B/TRPC7/GABRG2/KCNC1/KCNB2/SCN7A/KCNV1/SCNN1A/SCN4B/GRIA2/GRIA4/GRIK2/NLGN1/ABCC8/GRIN2A/GABRB3/SCN3B/SCN11A/CACNA1B/CLCNKB/GRIA1/CHRNA7/ANO2/KCNG3/DPP6/KCNA6/VWC2/DPP10/HTR3A/KCNA4/GRIK4/UNC80/GABRA3/GRIK1/LRRC18/CLCNKA/KCNMB2/KCNQ2/SHISA8/GRID2/HCN4/ATP4B/SCN1A/KCNK4/LRRC52/CATSPER4/VWC2L | 97 |
| GO:1990351 | GO:1990351 | transporter complex | 97/2609 | 332/19717 | 0 | 0 | 0 | KCNJ8/ABCG5/CHRNA4/TRPC5/ABCC9/OLFM2/ABCG8/CACNG1/KCNH4/FXYD2/CACNG8/CACNG4/GABRP/KCNE1B/KCNG4/CHRNA6/GLRA1/GABRR3/ATP1B4/KCNA10/CHRNB3/CHRND/ATP12A/GABRA6/BEST2/BEST4/SCNN1B/CNGA3/LRRC26/ATP1A2/GRIK3/KCNIP4/CHRNA3/TRPM4/HTR3B/OLFM3/CACNA2D2/SLC9A1/KCNH1/GRIK5/CNGB1/CLIC5/SCN3A/KCNK2/GLRA4/CASQ2/PLN/CACNA1A/GRIN1/KCNS3/SCN2B/TRPC7/GABRG2/KCNC1/KCNB2/SCN7A/KCNV1/SCNN1A/SCN4B/GRIA2/GRIA4/GRIK2/NLGN1/ABCC8/GRIN2A/GABRB3/SCN3B/SCN11A/CACNA1B/CLCNKB/GRIA1/CHRNA7/ANO2/KCNG3/DPP6/KCNA6/VWC2/DPP10/HTR3A/KCNA4/GRIK4/UNC80/GABRA3/GRIK1/LRRC18/CLCNKA/KCNMB2/KCNQ2/SHISA8/GRID2/HCN4/ATP4B/SCN1A/KCNK4/LRRC52/CATSPER4/VWC2L | 97 |
| GO:0034358 | GO:0034358 | plasma lipoprotein particle | 25/2609 | 37/19717 | 0 | 0 | 0 | APOA5/APOC4/APOC3/APOC2/APOA1/APOB/HPR/APOC1/APOA2/APOH/APOM/APOF/PON1/LIPC/SAA4/LPA/CLU/LCAT/APOE/SAA1/SAA2/CETP/MSR1/APOA4/APOBR | 25 |
Save data
Combine plots
Created and Maintained by Sangram Keshari Sahu
Rmarkdown Template used from Rmdplates package
Licensed under CC-BY 4.0
Source Code At GitHub